
CHAIN SPECIFICITY TRANSCRIPTOME ANALYSIS IN DIFFERENT TEMPERATURES CONDITIONED OF YERSINIA RUCKERI
-
摘要: 为鉴定鱼源鲁氏耶尔森氏菌(Yersinia ruckeri) SC09菌株水生环境中不同温度的转录组水平上的差异, 研究采用链特异性转录组测序(Strand-specific RNA-seq)技术对菌体生理温度(28℃)和实验培养温度(37℃)下进行链特异性测序, 原始数据质控后, 筛选得到差异表达基因, 通过KEGG (Kyoto Encyclopedia of Genes and Genomes)数据库对差异表达基因进行富集分析, 并利用Rockhopper软件筛选出的重要原核生物基因簇进行验证。结果显示, 共筛选获得173个显著差异表达基因(P<0.05), 其中包括58个上调基因, 主要富集到一些特殊的碳水化合物代谢相关的通路中; 以及115个下调基因, 主要富集到双组份信号系统中与三羧酸循环相关的代谢通路上, 同时部分基因富集到编码鞭毛素相关的基因簇中。结果表明, 相对于37℃的实验室培养温度, 在水生环境的生理温度条件下(28℃) SC09菌株拥有较高的运动性和较强的葡萄糖代谢, 但相对的SC09菌株代谢一些特殊糖类的能力减弱。Abstract: To investigate the effect of temperature on transcripts and relative mRNA levels, high-throughput RNA sequencing were used for Yersinia ruckeri grown at 28℃ and 37℃. After the quality control, all differentially regulated genes were enriched and annotated by Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and verified by Rockhopper software. The results showed that 173 differentially expressed genes were identified (P-value<0.05), including 58 up-regulated genes mainly enriched in several KEGG pathways: phosphotransferase system, starch and sucrose metabolism, galactose metabolism and other glycan degradation; and 115 down-regulated genes mainly enriched in several KEGG pathways: two-component system, thiamine metabolism and flagellar assembly. In summary, the SC09 had lower motility and weaker glucose metabolism at temperature of 37℃. The compensatory overexpression of some specific carbohydrate metabolism might play important roles for the survival of this bacterial under high temperature. This study provides an important basis for further research on the molecular mechanisms of SC09 in different temperatures.
-
鲁氏耶尔森氏菌(Yersinia ruckeri)主要引起鱼类肠炎红嘴病(Enteric redmouth disease, ERM)[1]。ERM是一种重要的水生动物疾病, 早于1966年美国爱德荷州的养殖虹鳟(Oncorhynchus mykiss)中暴发[1]。20世纪70年代至80年代由美国传至欧洲, 主要在英国和欧洲大陆之间传播, 宿主一般为在淡、海水中的野生鲑鱼和养殖鲑鱼[2]。然而, 自ERM 首次报道以来, 其宿主范围和地理分布逐渐扩大, 目前我国西南地区该病原被报道可感染养殖斑点叉尾鮰并引起严重死亡[3]。该病暴发往往由环境压力介导, 例如水温变化、水质差异等。本研究中心于2008年简阳三岔水库养殖斑点叉尾鮰(Ictalurus punctatus)暴发的细菌性败血症中分离得到的Y. ruckeri SC09株, 并随后进行了细菌基因组完成图测序[4, 5]。
病原菌引起的鱼类疾病暴发往往与水温有极为密切的相关性, 为进一步分析该病原在水生环境中的生理发病温度(28℃)和实验培养温度(37℃)条件下的基因转录水平的差异, 本实验进行了基于RNA-seq的菌体不同温度条件下的链特异性转录组分析。本实验期望通过高通量测序技术更全面了解菌体中生物信息的变化情况, 为研究相关差异基因开创新的方法和思路, 也为进一步研究Y. ruckeri SC09菌株的致病机制和生存机制提供更丰富数据参考。
1. 材料与方法
1.1 菌株
本研究中测序和实验所用菌株为2008年简阳三岔水库养殖斑点叉尾鮰(Ictalurus punctatus)暴发的细菌性败血症中分离得到的鲁氏耶尔森氏菌(Yersinia ruckeri)的SC09株, 由四川农业大学鱼病研究中心分离保存。该菌株已进行了细菌基因组完成图测序, 菌株染色体NCBI基因组登录号为: NZ_CP025800.1; 该菌株还含有2个野生型质粒, 分别命名为pLT和pWKY, 其NCBI登录号分别为: NZ_CP025802.1和NZ_CP025801.1。
1.2 不同温度条件下菌株培养及其总RNA提取
为了研究菌株Y. ruckeri SC09在常规实验室菌体培养温度(37℃)和该菌株分离时宿主所在环境的发病温度(28℃)条件下菌体基因表达的差异, 本研究将菌株SC09在LB培养基(北京欣经科生物技术有限公司)上划线涂板, 置于37℃培养16h后, 挑取单菌落转接至10 mL LB培养基, 分别在37℃(样本命名为SC09-37)和28℃(样本命名为SC09-28)180 r/min 过夜培养A600至0.8—0.9。离心收集菌体, 按照TRIzol® Reagent提取试剂盒(Invitrogen)操作说明进行菌体的总RNA 抽提, 并电泳质检合格后纯化总RNA。
1.3 构建链特异性测序文库并高通量测序
采用Illumina TruseqTM RNA sample prep Kit试剂(Invitrogen公司)并以5 μg total RNA起始量建库; 磁珠法除rRNA(Ribo-Zero Magnetic kit (G+/G-Bacteria), EpiCentre公司); 离子打断mRNA(TruseqTM RNA sample prep Kit, Illumina公司), 双链cDNA合成, 第二链合成时采用dUTP代替dTTP, 并在合成双链后接index接头(TruseqTM RNA sample prep Kit, Illumina公司), 加入UNG酶(Illumina公司)降解cDNA第二链; 文库富集, PCR扩增15个cycles; 2%琼脂糖胶回收目的条带(Certified Low Range Ultra Agarose, Bio-Rad公司); TBS380(Picogreen)定量, 按数据比例混合上机; cBot(仪器产自Illumina公司)桥式PCR扩增得到clusters; Hiseq(仪器产自Illumina公司)平台进行2×150 bp双端测序。
1.4 原始数据统计和质控
对所有测序reads的每个circle进行碱基分布和质量波动统计, 得到(Raw data), 宏观反映测序质量和文库质量。Illumina Hiseq原始数据中会包含接头序列、低质量reads、N率较高序列及长度过短序列, 其将影响后续序列组装。为保证后续分析准确性, 首先对原始测序数据进行过滤, 从而得到高质量的测序数据(Clean data)以保证后续分析的顺利进行, 具体步骤如下: 去除reads中的接头序列, 去除由于接头自连等原因导致没有插入片段的reads; 将序列末端(3′端)质量较低(质量值小于20)的碱基修剪掉, 如剩余序列中仍然有质量值小于10的碱基, 则将整条序列剔除, 否则保留; 去除含N比率超过10%的reads; 舍弃去adapter及质量修剪后长度小于70 bp的序列。质控使用软件: SeqPrep (https://github.com/jstjohn/SeqPrep)和Sickle (https://github.com/najoshi/sickle)。
1.5 与参考基因组比对
利用HTseq-count软件并根据reads比对结果分配reads到特定的转录本进行转录本reads计算分析(Read count)。采用Rockhopper软件将各样品过滤后的测序序列与参考基因组进行比对, 使其定到基因组。使用IGV(Integrative Genomics Viewer)浏览器对测序的bam文件格式化后的wig文件进行可视化浏览以便于在不同尺度上显示基因不同区域的丰度, 以反映不同区域的转录水平。
1.6 基因表达定量和宏观差异分析
针对Read counts结果进一步采用FPKM (Fragments Per Kilobase of transcript per Million fragments mapped)来表征基因表达量。
本研究菌体样本无生物学重复, 一般用logFC(FC是fold change的简写, logFC是FC的对数值, 意为“差值倍数”)来对Read counts值进行过滤, 随后利用DESeq读入, 通过在DESeq软件内部进行标准化后进行MA图的绘制。
1.7 差异表达基因的KEGG富集分析
本次分析使用KOBAS (http://kobas.cbi.pku.edu.cn/home.do)进行KEGG PATHWAY富集分析, 使用Fisher精确检验进行计算。为控制计算假阳性率, 采用BH (FDR)方法进行多重检验, 计算公式与上节相同, 经过校正的P (Corrected P-Value)以0.05为阈值, 满足此条件的KEGG通路定义为在差异表达基因中显著富集的KEGG通路。
1.8 毒力相关基因簇的操纵子分析
结合RNA-seq实验数据(计算所得基因表达量), 采用Rockhopper软件(http://cs.wellesley.edu/~btjaden/Rockhopper/)预测操纵子。具体是联合基因间距离和基因表达量相关性2个特征用朴素贝叶斯分类器模型来预测操纵子, 该预测算法的敏感性和特异性均高达95%。同时, 对预测得到操纵子进行长度分布、包含的结构基因数目和操纵子链分布进行计算。进一步地, 从预测到的操纵子中进行毒力相关性分析。
2. 结果
2.1 原始数据统计和原始数据质控
Illumina测序得到的原始图像数据经过Base Calling转化为序列数据, 结果以FASTQ文件格式来存储。FASTQ文件为最原始的数据文件, 文件包含测序read的序列信息以及测序质量信息。对SC09-28和SC09-37两个样本的所有原始reads进行统计, 得到所有原始读长的数量(Raw reads)和碱基总数(Raw base), 统计碱基的Quality scores (Q20和Q30), 具体结果见表 1。
表 1 原始数据统计Table 1. Raw data statistics样本名称Sample 原始读长Raw reads 原始总碱基数Raw base Q20质控Q20 (%) Q30质控Q30 (%) SC09-28 46412652 6961897800 98.34 95.42 SC09-37 31779760 4766964000 98.05 94.51 注: Q值指测序过程碱基识别过程中, 对所识别的碱基给出的错误概率; Q20. 错误识别概率是1%, 正确率是99%; Q30. 错误识别概率是0.1%, 正确率是99.9%; 下同
Note: The Q value is the error probability given to the identified base during the base recognition process. Q20: The error recognition probability is 1%, and the correct rate is 99%; Q30: the error recognition probability is 0.1%, and the correct rate is 99.9%; the same applies below原始数据中包括测序接头序列、低质量读段、N率较高序列及长度过短序列, 经过过滤后得到质量更好的数据。原始数据质控后得到的具体结果见表 2。
表 2 原始数据质控Table 2. Raw data quality control样本名称Sample 原始读长Clean reads 原始总碱基数Clean base Q20质控Q20 (%) Q30质控Q30 (%) SC09-28 43949712 6560903356 99.02 96.75 SC09-37 29528546 4392474370 98.78 95.89 2.2 参考基因组比对
SC09-28和SC09-37两个样品过滤后的测序序列分别与参考基因组(包括pLT和pWKY两个质粒的参考基序列)进行比对, 使其定位到基因组。采用Rockhopper软件进行比对, Rockhopper是一个全面的主要针对原核转录组数据的计算分析软件, 采用的比对算法类似Bowtie2, 基于BWT (Burrows-Wheeler_transform)数据转化算法构建参考基因组的FM-index, 使比对更加准确快速。比对得到的结果如表 3所示。
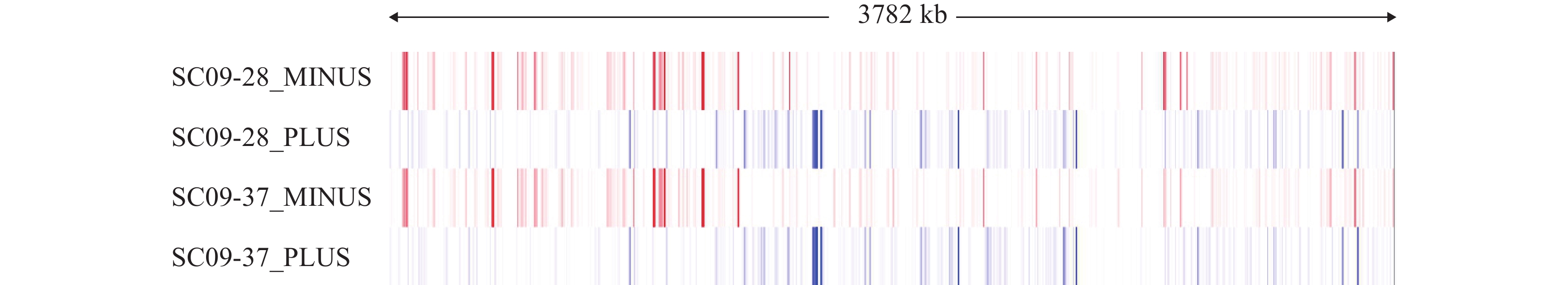
表 3 原始数据比对到基因组Table 3. The raw data mapped to the genome样本Sample 总reads数Total reads 匹配上的reads数Mapped reads 匹配比例Mapped ratio (%) 参考基因组Reference genome SC09-28 23206326 22209667 96 Y. ruckeri SC09 chromosome SC09-28 23206326 41191 0.17 Y. ruckeri SC09 plasmid pLT SC09-28 23206326 45196 0.19 Y. ruckeri SC09 plasmid pWKY SC09-37 15889880 14672184 92 Y. ruckeri SC09 chromosome SC09-37 15889880 28176 0.17 Y. ruckeri SC09 plasmid pLT SC09-37 15889880 28719 0.18 Y. ruckeri SC09 plasmid pWKY 同时, 利用 RNA-seq reads在unigene上比对结果的wig格式文件, 使用IGV (Integrative Genomics Viewer)浏览器对wig文件进行可视化浏览, 通过IGV在不同尺度上显示基因不同区域的丰度, 以反映不同区域的转录水平, 结果如图 1所示。SC09-28_MINUS是指样本SC09-28 (28℃)的负义链基因的表达reads比对到SC09基因组的贴合水平,SC09-28_PLUS是指样本SC09-28 (28℃)的正义链基因的表达reads比对到SC09基因组的贴合水平,SC09-37_MINUS是指样本SC09-37(37℃)的负义链基因的表达reads比对到SC09基因组的贴合水平, SC09-37_PLUS是指样本SC09-37(37℃)的正义链基因的表达reads比对到SC09基因组的贴合水平。
2.3 差异表达基因宏观分析
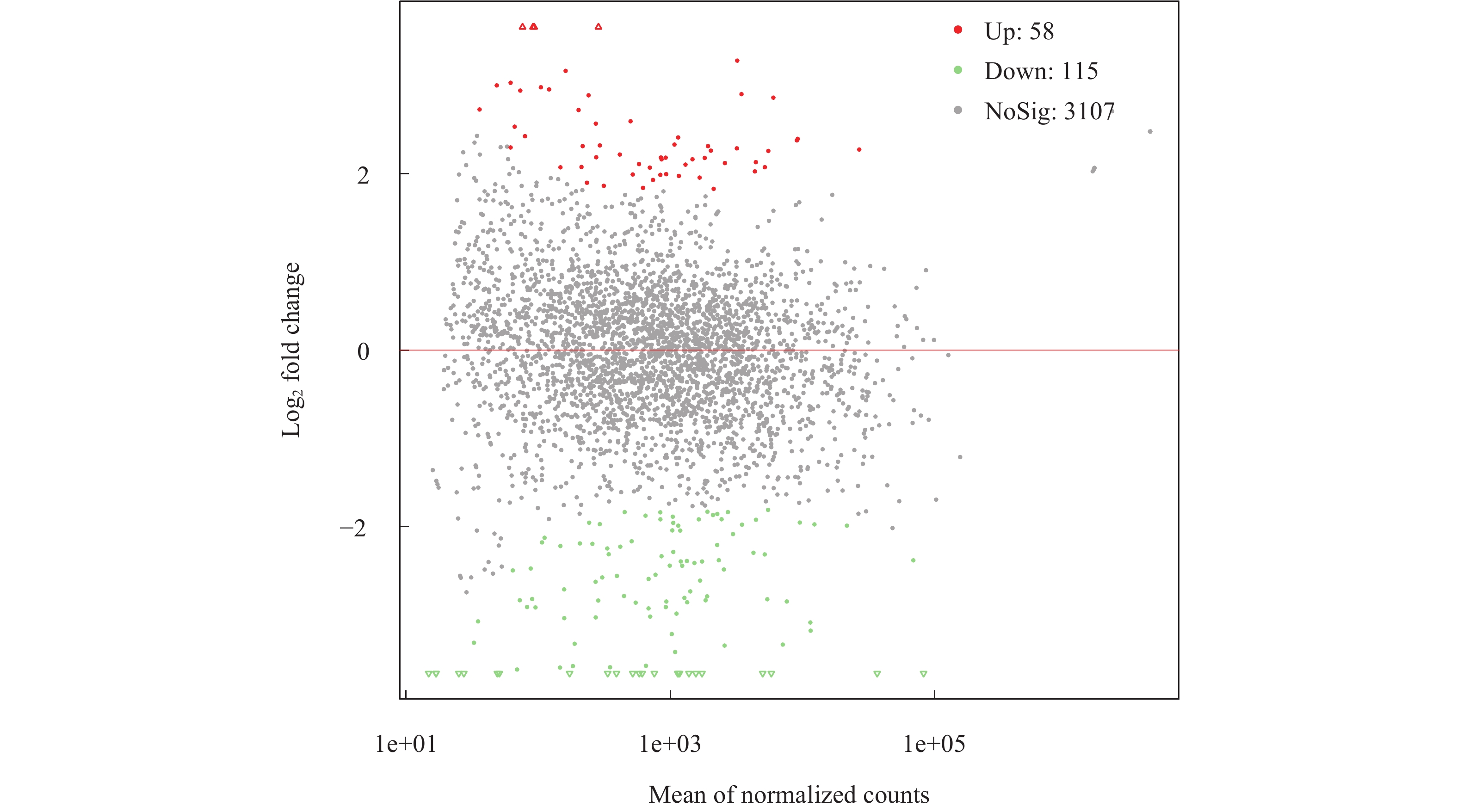
对Gene差异表达筛选结果进行汇总统计如下, 比较P<0.05阈值条件下的显著上调和显著下调的基因, 并绘制MA图(图 2)。图中X轴为标准化后序列计数在两组间的平均数, Y轴代表倍数变化的对数值。从X轴看, 从左往右代表基因表达量从低到高; 从Y轴看, 越偏离Y=0这条曲线则代表倍数变化越大。位于红色直线上方的为表达上调基因, 位于红线下方的为表达下调的基因。红色高亮数据点代表符合特定筛选阈值条件的显著表达上调基因。绿色高亮数据点代表符合特定筛选阈值条件的显著表达下调基因。由图可知, 样本SC09-37相对于SC09-28, 其显著上调的基因有58个, 而显著下调的基因有115个, 有差异表达但不显著(不满足阈值条件)的基因有3107个。
![]() 图 2 SC09差异基因筛选MA图(SC09-37相对于SC09-28)Figure 2. MA plot of differentially expressed genes in SC09 (SC09-37 compare to SC09-28)
图 2 SC09差异基因筛选MA图(SC09-37相对于SC09-28)Figure 2. MA plot of differentially expressed genes in SC09 (SC09-37 compare to SC09-28)2.4 差异表达基因的KEGG富集分析
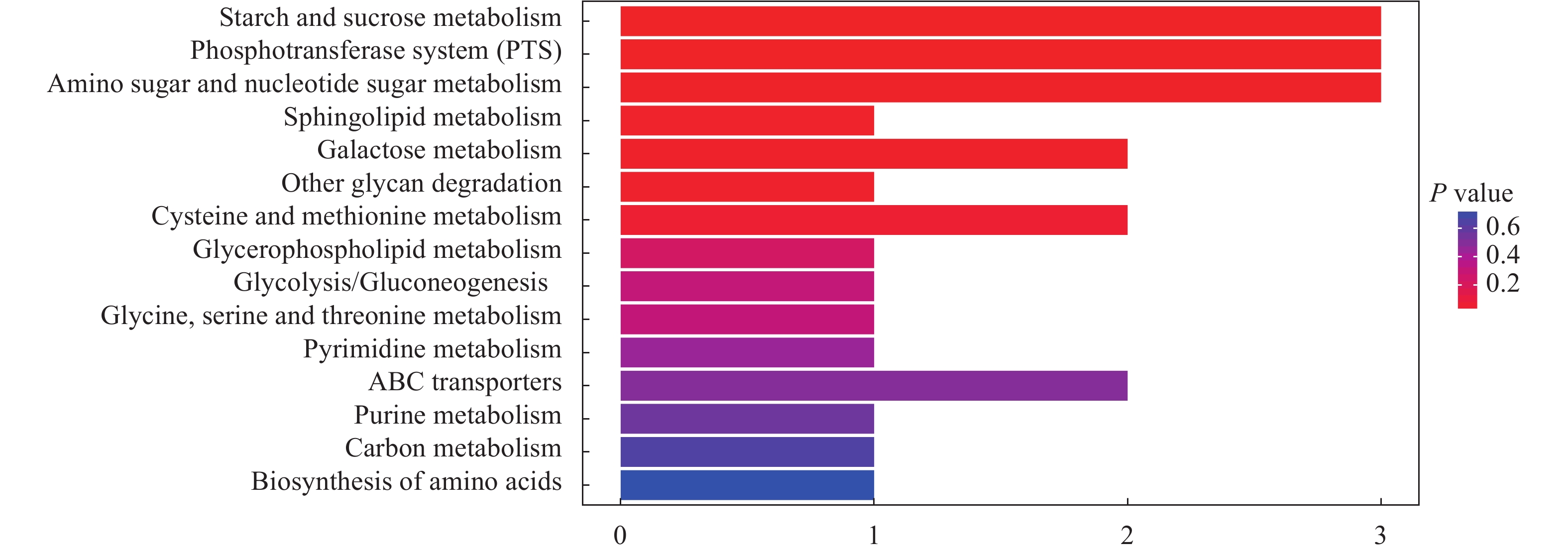
将2个样本的P<0.05的差异基因进行KEGG功能富集分析(图 3和图 4)。SC09-37相对于SC09-28共有58个基因显著上调表达(图 2), 总计可印射到15个KEGG的pathway条目, 经过超几何分布检验进行统计计算后, 再次按阈值P<0.05进行过滤后得到7个显著富集的KEGG条目(图 3, 红色条目)。这些显著上调的基因几乎都与各种糖类的获取和代谢相关, 包括磷酸转移酶系统(Phosphotransferase system, PTS)相关的基因、淀粉和蔗糖代谢(Starch and sucrose metabolism)相关基因、氨基糖和核糖(Amino sugar and nucleotide sugar metabolism)相关基因、半乳糖代谢(Galactose metabolism)相关基因、其他多糖代谢(Other glycan degradation)相关基因等。此外, 也包括较少的与脂类(Sphingolipid metabolism)和氨基酸(Cysteine and methionine metabolism)代谢相关基因。由此可知, SC09菌株在温度较高的37℃时, 其糖类代谢相关基因呈现显著上调表达。需要注意的是这些显著上调的KEGG的pathway中有重复出现的基因, 其中 PTS相关的基因与Starch and sucrose metabolism相关有基因部分重复。二者都包含了NJ56_RS13690(NCBI登录号)和NJ56_RS13695 (NCBI登录号)两个基因。
![]() 图 3 上调表达基因的KEGG富集分析(SC09-37相对于SC09-28)Figure 3. KEGG enrichment analysis of up-regulated genes (SC09-37 compare to SC09-28)
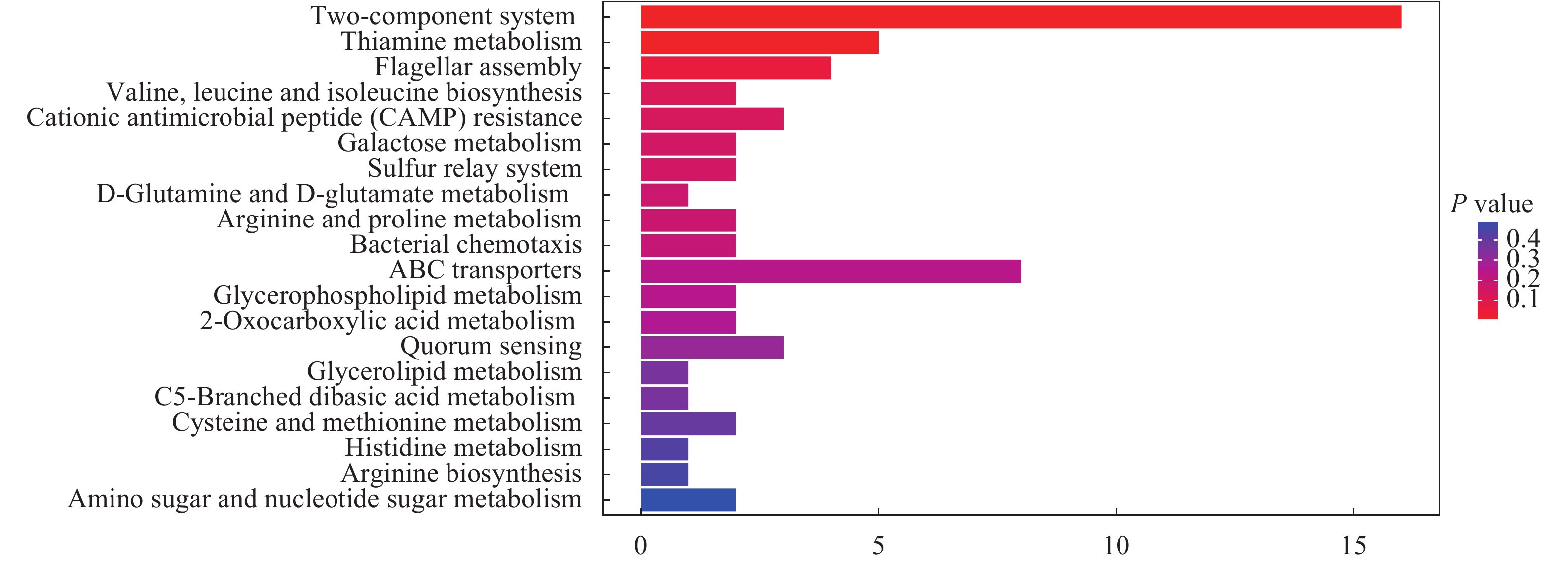
图 3 上调表达基因的KEGG富集分析(SC09-37相对于SC09-28)Figure 3. KEGG enrichment analysis of up-regulated genes (SC09-37 compare to SC09-28)![]() 图 4 下调表达基因的KEGG富集分析(SC09-37相对于SC09-28)Figure 4. KEGG enrichment analysis of down-regulated genes (SC09-37 compare to SC09-28)
图 4 下调表达基因的KEGG富集分析(SC09-37相对于SC09-28)Figure 4. KEGG enrichment analysis of down-regulated genes (SC09-37 compare to SC09-28)This page contains the following errors:
error on line 1 at column 1: Start tag expected, '<' not foundBelow is a rendering of the page up to the first error.
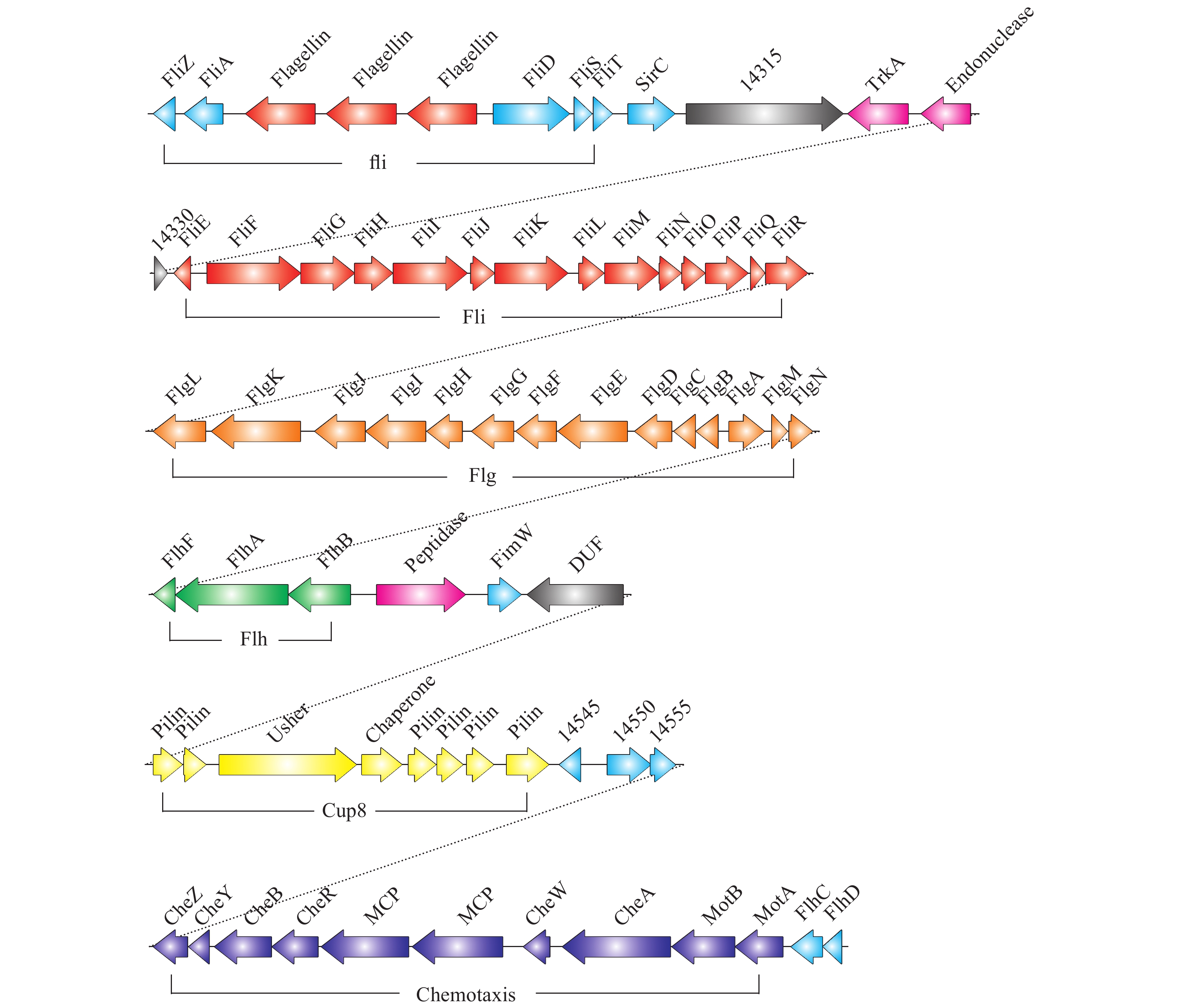
2.5 毒力相关基因簇的鞭毛系统操纵子分析
基因原核链特异性转录组本研究预测了Y. ruckeri SC09鞭毛相关系统的完整操纵子, 包括鞭毛主体、调控和趋化三大系统(图 5)。Y. ruckeri SC09带有完整的鞭毛主体成分基因簇fli (NJ56_ 14270-NJ56_14400, 橘红色基因簇)、flg (NJ56_ 14405-NJ56_14470, 金黄色基因簇)、flh (NJ56_ 14475-NJ56_14485, 绿色基因簇), 其后紧随着调控鞭毛旋转的趋化系统(NJ56_14560-NJ56_14605)以及启动装配鞭毛主体结构的一级调控基因flhC (NJ56_14610)、flhD (NJ56_14615)。比较特殊的是, 在fli-flg-flh基因簇和趋化系统之间还存在着一个编码 CUP (Chaperone-usher pathway)型菌毛的基因簇(命名为 cup8, NJ56_14495-NJ56_14540), 推测其可能有助于鞭毛的协同运动。
3. 讨论
3.1 原核生物转录组研究特性
RNA-seq的测序质量分析是评估转录组当中非常重要的数据内容, 但需要注意的是一般的真核生物的转录组测序与细菌等原核生物转录组测序的测序策略具有明显的差异。真核生物由于具有polyA尾巴, 所以可以采用polyA募集mRNA, 进而展开cDNA双链的合成。因此, 真核生物转录组在测序序列中往往不能提供链方向的信息, 难以确定反义转录本, 且不能真实的反映转录情况。原核生物由于不具有polyA尾巴, 所以采取了链特异性建库策略。此方法是在构建文库时, 利用高保真Taq酶将mRNA链的方向信息存于测序文库中, 一般最终数据分析可判断转录本是来自正义还是负义链。原核生物转租组测序策略目前仅能采用链特异性的方法。
测序质量的好坏将直接影响后续分析的准确性[6]。由表 1和表 2可知, 本研究2个样本原始数据的Q20和Q30处于非常高的水平, 而进一步经过质控后2个样本的Q20更是高达99.02%和98.78%, Q30也高达96.75%和95.89%。同时, 由表 3和图 3可知原始数据匹配到SC09染色体基因组的比率也高达96%和92%。由此可知, 本研究测序数据准确性高且基因组覆盖率完整, 在此基础上进行的差异分析和基因富集分析以及操纵子基因簇分析十分可靠。
3.2 鲁氏耶尔森菌SC09宏观差异分析
基因表达具有时间和空间特异性, 外界刺激和内部环境都会影响基因的表达。在两个不同条件下, 表达水平存在显著差异的基因, 称之为差异表达基因[7]。在生物信息学中, 寻找差异表达基因的过程叫做差异表达分析, 也是本研究中RNA-seq分析的核心内容。在本研究中, P<0.05时, SC09-37相对于SC09-28显著上调的基因有58个, 而显著下调的基因有115个。宏观来看, 在较高的实验室培养温度下(37℃)菌株多数基因明显处于下调状态, 菌株相对显得不那么“活跃”, 而较低的温度下(28℃)菌株多数基因将显著上调, 菌株表现出更为“活跃”的状态。
3.3 菌株基本代谢与温度相关性
具体地, 通过基因的KEGG富集分析来看, 菌株SC09在温度较高时(37℃)双组份信号系统通路[8]中编码柠檬酸合酶(Citrate lyase)相关的基因(NJ56_RS12650、NJ56_RS12655、NJ56_RS12660、NJ56_RS12665和NJ56_RS12670)显著下调, 这将导致菌株的三羧酸循环被抑制, 代谢葡萄糖的能力显著降低, 反之, 28℃时菌株具有较好的代谢葡萄糖的能力。同时, 硫胺素(维生素B1, VB1)代谢通路相关基因NJ56_RS04720、NJ56_RS04725、NJ56_RS04730、NJ56_RS04740和NJ56_RS04745在高温时显著下调。VB1在菌体内主要以焦磷酸硫胺素(TPP)形式存在, 是α-酮酸氧化脱羧酶复合物的重要辅酶[9]。高温时菌株相应的辅酶的功能的降低, 进一步抑制了菌株的三羧酸循环。这些基因的变化趋势表明耶尔森氏菌更加趋向于在较低水温条件下生存, 且在较低温度下菌体往往表现出更加活跃的代谢及其他生命活动。这进一步提示菌体引发鱼类宿主疾病往往与其所在水生环境温度密切相关。
3.4 菌株鞭毛系统转录分析
由于原核生物是多顺反子mRNA, 具有5′、3′UTR, 不具有poly(A)尾巴, 转录与翻译同时进行, 具有操纵子结构(很长的基因簇), 所以我们通过RNA-seq完整的描绘了SC09菌株的编码鞭毛相关的基因簇。细菌逆化学梯度的游动能力有赖于于马达驱动的鞭毛(Flagellum), 其可通过跨膜离子运动(Proton motive force, PMF)所提供的能量来带动鞭毛的丝状纤维的旋转[10]。鞭毛也是一个分层基因调控的典范, 其可通过蛋白间互作和对蛋白分泌的控制在较短时间和较小空间中细致精密地装配成一个复杂的多蛋白结构[11]。因为鞭毛的重要性, 植物和动物会专一性地产生识别鞭毛的受体来介导天然免疫反应[12], 同时, 作为一种精细自组装的纳米机器, 它使得细菌能向更好的生活环境运动, 在细菌的生命活动中也发挥了重要作用(包括运动性、 致病性等)[13]。细菌的鞭毛呈长细丝状并突出于菌体表面, 其结构主要由三部分组成: 引擎(Engine)、鞭毛丝(Propeller)以及连接引擎和鞭毛丝的接头部分(Universal joint)[14]。此外, 细菌利用趋化性(Chemotaxis)向对它们生长有利的环境移动, 其表面的化学受体会侦测环境信号的变化并通过双组份系统(Two-component systems)控制鞭毛的旋转方向, 从而调节细菌的泳动方向[15], 所以细菌的趋化系统一般与细菌的鞭毛系统毗邻。同时, 鞭毛的组装还是一个多级反应, 因此调控鞭毛组装的多个调控基因往往也靠近鞭毛系统[16]。菌株SC09在温度较高时, 编码鞭毛素(Flagellin FliC)的基因NJ56_RS13110、NJ56_RS13115和NJ56_RS13120也显著下调, 意味着菌体鞭毛的主体部分表达的降低或不表达, 提示菌体在实验室条件下(37℃)运动性不如在一般水体环境中(28℃)那么强烈。这表明, 较高温度时, SC09菌株的运动能力较弱, 游泳性较差。
3.5 菌株碳源摄取能力与基因转录水平分析
菌株SC09在温度较高时上调表达的基因中多数显著富集到一些特殊的碳水化合物(糖类)代谢相关的pathway: 磷酸转移酶系统(Phosphotransferase system, PTS)[17]、淀粉和蔗糖代谢(Starch and sucrose metabolism)、氨基糖和核糖(Amino sugar and nucleotide sugar metabolism)、半乳糖代谢(Galactose metabolism) 等。这表明, 尽管温度较高时, SC09菌株葡萄糖代谢通路受阻, 但代偿性地提高了其他碳水化合物相关基因的表达。这提示在实验室培养条件下(37℃)SC09菌株往往不是以葡萄糖作为其主要摄入碳源, 而是以更为复杂多样的其他碳水化合物来代替葡萄糖作为其碳源。这些基因变化趋势进一步说明了耶尔森氏菌体的高温适应性机制, 这为细菌在某一水生环境中长期生存提供了生理基础。
本研究首次揭示了Y. ruckeri SC09菌株在水生环境中的生理发病温度(28℃)和实验培养温度(37℃)条件下的基因转录水平的差异, 通过高通量测序技术更全面揭示了菌体中生物信息的变化情况, 为进一步研究Y. ruckeri SC09菌株的致病机制和生存机制提供了数据参考。
-
![]()
图 2 SC09差异基因筛选MA图(SC09-37相对于SC09-28)
Figure 2. MA plot of differentially expressed genes in SC09 (SC09-37 compare to SC09-28)
![]()
图 3 上调表达基因的KEGG富集分析(SC09-37相对于SC09-28)
Figure 3. KEGG enrichment analysis of up-regulated genes (SC09-37 compare to SC09-28)
![]()
图 4 下调表达基因的KEGG富集分析(SC09-37相对于SC09-28)
Figure 4. KEGG enrichment analysis of down-regulated genes (SC09-37 compare to SC09-28)
表 1 原始数据统计
Table 1 Raw data statistics
样本名称Sample 原始读长Raw reads 原始总碱基数Raw base Q20质控Q20 (%) Q30质控Q30 (%) SC09-28 46412652 6961897800 98.34 95.42 SC09-37 31779760 4766964000 98.05 94.51 注: Q值指测序过程碱基识别过程中, 对所识别的碱基给出的错误概率; Q20. 错误识别概率是1%, 正确率是99%; Q30. 错误识别概率是0.1%, 正确率是99.9%; 下同
Note: The Q value is the error probability given to the identified base during the base recognition process. Q20: The error recognition probability is 1%, and the correct rate is 99%; Q30: the error recognition probability is 0.1%, and the correct rate is 99.9%; the same applies below 下载: 导出CSV
下载: 导出CSV
表 2 原始数据质控
Table 2 Raw data quality control
样本名称Sample 原始读长Clean reads 原始总碱基数Clean base Q20质控Q20 (%) Q30质控Q30 (%) SC09-28 43949712 6560903356 99.02 96.75 SC09-37 29528546 4392474370 98.78 95.89
下载: 导出CSV
表 3 原始数据比对到基因组
Table 3 The raw data mapped to the genome
样本Sample 总reads数Total reads 匹配上的reads数Mapped reads 匹配比例Mapped ratio (%) 参考基因组Reference genome SC09-28 23206326 22209667 96 Y. ruckeri SC09 chromosome SC09-28 23206326 41191 0.17 Y. ruckeri SC09 plasmid pLT SC09-28 23206326 45196 0.19 Y. ruckeri SC09 plasmid pWKY SC09-37 15889880 14672184 92 Y. ruckeri SC09 chromosome SC09-37 15889880 28176 0.17 Y. ruckeri SC09 plasmid pLT SC09-37 15889880 28719 0.18 Y. ruckeri SC09 plasmid pWKY
下载: 导出CSV
-
[1] Kumar G, Menanteau-Ledouble S, Saleh M, et al. Yersi-nia ruckeri, the causative agent of enteric redmouth di-sease in fish [J]. Veterinary Research, 2015, 46(1): 103 doi: 10.1186/s13567-015-0238-4
[2] 杨移斌, 夏永涛, 郑卫东, 等. 鲟源鲁氏耶尔森氏菌的分离鉴定及药敏特性研究. 水生生物学报, 2013, 37(2): 393—398 doi: 10.7541/2013.33 Yang Y B, Xian L Y, Zhang W D, et al. Isolation and identification of Yersinia ruckeri from Acipenser baerii and its antibiotic sensitivity [J]. Acta Hydrobiologica Sini-ca, 2013, 37(2): 393—398 doi: 10.7541/2013.33
[3] 阳磊, 汪开毓, 周燕, 等. 鲁氏耶尔森氏菌口服微球疫苗特性分析及免疫效果研究. 水生生物学报, 2015, 39(6): 1142—1149 doi: 10.7541/2015.150 Yang L, Wang K Y, Zhou Y, et al. The characteristics and immune efficacy of an oral microspheres vaccine of Yersinia ruckeri [J]. Acta Hydrobiologica Sinica, 2015, 39(6): 1142—1149 doi: 10.7541/2015.150
[4] Wang K Y, Liu T, Wang J, et al. Complete genome sequence of the fish pathogen Yersinia ruckeri strain SC09, isolated from diseased Ictalurus punctatus in China [J]. Genome Announc, 2015, 3(1): 1—2
[5] Tao L, Wang K Y, Wang J, et al. Genome sequence of the fish pathogen Yersinia ruckeri SC09 provides insights into niche adaptation and pathogenic mechanism [J]. International Journal of Molecular Sciences, 2016, 17(4): 557 doi: 10.3390/ijms17040557
[6] Tjaden B. De novo assembly of bacterial transcriptomes from RNA-seq data [J]. Genome Biology, 2015, 16(1): 1 doi: 10.1186/s13059-014-0572-2
[7] Levin J Z, Yassour M, Xian A, et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods [J]. Nature Methods, 2010, 7(9): 709—715 doi: 10.1038/nmeth.1491
[8] Mitrophanov A Y, Groisman E A. Signal integration in bacterial two-component regulatory systems [J]. Genes & Development, 2008, 22(19): 2601
[9] Zhu H, Sun G, Zhu B. An investigation report about the lack of VB1 [J]. Journal of Thoracic Disease, 2016, 8(8): 2244—2246 doi: 10.21037/jtd
[10] Anderson J K, Smith T G, Hoover T R. Sense and sensibi-lity: flagellum-mediated gene regulation [J]. Trends in Microbiology, 2010, 18(1): 30—37 doi: 10.1016/j.tim.2009.11.001
[11] Maki-Yonekura S, Yonekura K, Namba K. Conformational change of flagellin for polymorphic supercoiling of the flagellar filament [J]. Nature Structural & Molecular Biology, 2010, 17(4): 417
[12] Belas R. Biofilms, flagella, and mechanosensing of surfaces by bacteria [J]. Trends in Microbiology, 2014, 22(9): 517—527 doi: 10.1016/j.tim.2014.05.002
[13] Porter S L, Wadhams G H, Armitage J P. Signal processing in complex chemotaxis pathways [J]. Nature Reviews Microbiology, 2011, 9(3): 153—165 doi: 10.1038/nrmicro2505
[14] Beatson S A, Minamino T, Pallen M J. Variation in bacterial flagellins: from sequence to structure [J]. Trends in Microbiology, 2006, 14(4): 151 doi: 10.1016/j.tim.2006.02.008
[15] Partridge J D, Harshey R M. Swarming: flexible roaming plans [J]. Journal of Bacteriology, 2013, 195(5): 909—918 doi: 10.1128/JB.02063-12
[16] Stevenson E, Minton N P, Kuehne S A. The role of flagella in Clostridium difficile pathogenicity [J]. Trends in Microbiology, 2015, 23(5): 275 doi: 10.1016/j.tim.2015.01.004
[17] Deutscher J, Francke C, Postma P W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria [J]. Microbiology and Molecular Biology Reviews, 2006, 70(4): 939—1031 doi: 10.1128/MMBR.00024-06
-
期刊类型引用(5)
1. 贾新蕾,黄增朝,杨林狄,吕静,李妍萍,简纪常,黄郁葱. 不同培养温度的鱼源海豚链球菌转录组分析. 热带生物学报. 2024(01): 109-121 .  百度学术
百度学术
2. 刘艳,谷福蝶,陈慧莹,李焱,周钰,张军,刘红,曹敏杰,刘光明,刘庆梅. 基于RBL-2H3模型分析过敏反应效应细胞激活的差异基因. 食品科学. 2022(22): 105-112 . 百度学术
3. 张杰,尹强,闫志坚,万永青,王育青. 硅对干旱胁迫下燕麦根系响应的分子机理. 分子植物育种. 2021(14): 4759-4769 . 百度学术
4. 裴迪,刘志明,胡碧茹,吴文健. 基于不同氮源培养条件的巴氏芽孢杆菌脲酶功能转录组分析. 生物化学与生物物理进展. 2021(09): 1063-1076 . 百度学术
5. 王磊,邹清华,张慧芳,张建中,肖迪. 胃黏膜相关淋巴组织淋巴瘤幽门螺杆菌分离株与胃炎分离株的转录组比较. 疾病监测. 2020(04): 288-293 . 百度学术
其他类型引用(4)
计量
- 文章访问数: 8502
- HTML全文浏览量: 6548
- PDF下载量: 48
- 被引次数: 9